¡Atención!
Estudio Computacional
Las proteasas de coronavirus son objetivos atractivos para el diseño de medicamentos antivirales.
En este mundo de viajes rápidos y fáciles, los virus emergentes se están convirtiendo cada vez más en un gran peligro para la salud mundial. Los coronavirus son un ejemplo notable. Formas particularmente virulentas han surgido de sus huéspedes animales naturales y representan una amenaza para las comunidades humanas. En 2003, el virus del SARS surgió en China de las poblaciones de murciélagos, pasando a las civetas y finalmente a los humanos. Diez años después, el virus MERS también emergió de los murciélagos, transfiriéndose en el Medio Oriente a camellos dromedarios y luego a humanos. Recientemente, otro coronavirus ha surgido en China a través de animales en un mercado vivo. La biología estructural nos está ayudando a comprender a estos peligrosos enemigos y, con suerte, nos ayudará a desarrollar nuevas formas de combatirlos.
Las proteasas son enzimas que digieren las proteínas. Grupo de enzimas que hidrolizan las proteínas transformándolas en aminoácidos o en péptidos sencillos. Las proteasas son enzimas que rompen los enlaces peptídicos de las proteínas. Usan una molécula de agua para hacerlo y por lo tanto se clasifican como hidrolasas.
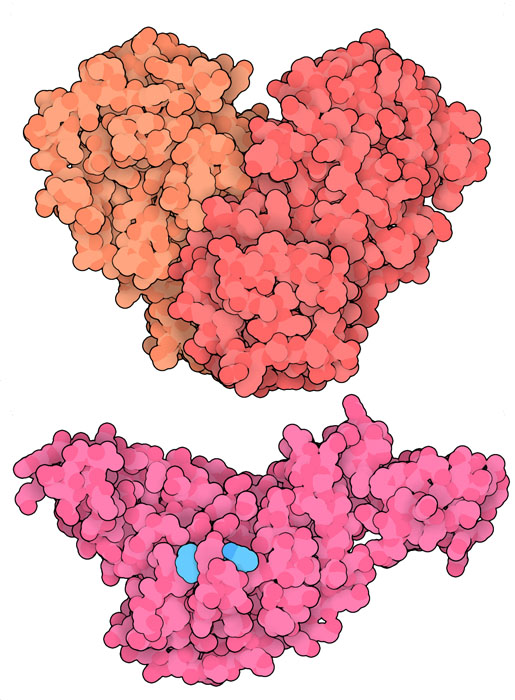
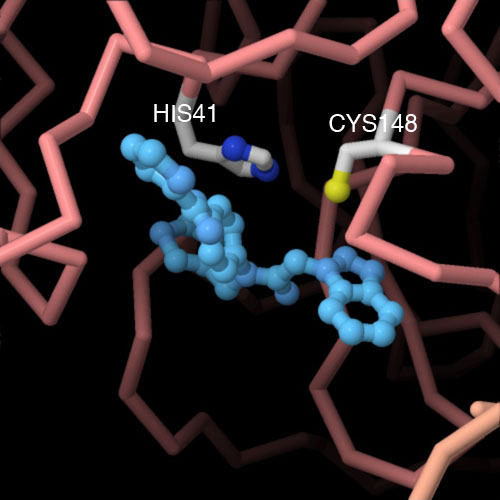
La estructura cristalina de alta resolución del coronavirus COVID-19 fue determinada por Zihe Rao y el equipo investigación de Haitao Yang en la Universidad Tecnológica de Shangai. Su publicación fue hecha el 5 de febrero de 2020 en la base de datos Protein Data Bank. La principal proteasa del coronavirus realiza la mayoría de estos cortes. El que se muestra aquí (entrada PDB 6lu7) es del coronavirus SARS-CoV-2 (2019-nCoV) que actualmente representa un peligro en Wuhan. Es un dímero de dos subunidades idénticas que juntas forman dos sitios activos. El pliegue proteico es similar a las serina proteasas como la tripsina, pero un aminoácido cisteína y una histidina cercana realizan la reacción de corte proteico y un dominio adicional estabiliza el dímero. Esta estructura tiene un inhibidor similar a un péptido unido en el sitio activo.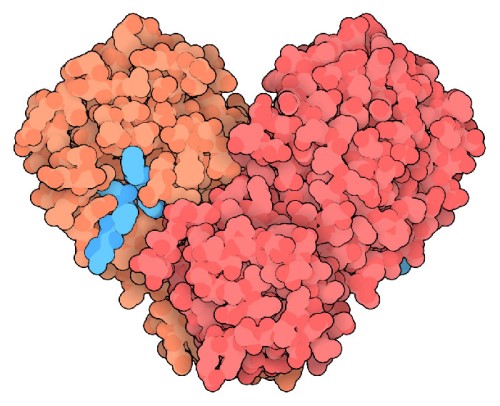
Ver archivo PDB en el simulador 3D
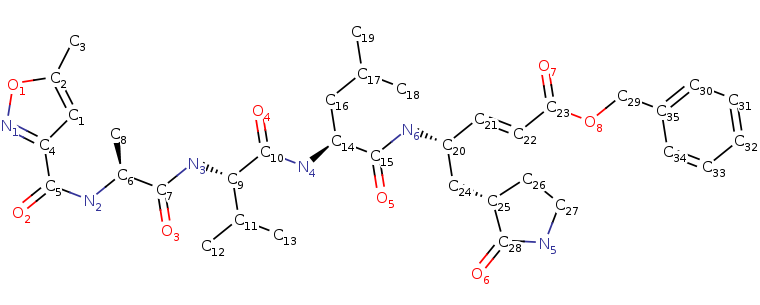
Inhibidor tipo péptido.
Ver archivo PDB tipo péptido en el simulador 3D
Ver archivo del SARS PDB en el simulador 3D
Ver archivo del SARS PDB en el simulador 3D
Ver archivo PDB en el simulador 3D.
Ver el sitio activo del archivo PDB con N-acetil-D-glucosamina en el simulador 3D.
De acuerdo a la publicación del 19 de febrero de 2020 en la revista Science, el brote de un nuevo betacoronavirus (2019-nCoV) representa una amenaza pandémica que ha sido declarada una emergencia de salud pública de preocupación internacional. La glicoproteína de espiga del CoV (S) es un objetivo clave para hacer una vacuna, anticuerpos terapéuticos y diagnósticos. Para facilitar el desarrollo de la contramedida médica (MCM), determinamos una estructura de crio microscopía electrónica de resolución de 3,5 Å del trímero S 2019-nCoV en la conformación de prefusión. El estado predominante del trímero tiene uno de los tres dominios de unión al receptor (RBD) rotados hacia arriba en una conformación accesible al receptor. También mostramos evidencia biofísica y estructural de que el 2019-nCoV S se une a ACE2 con mayor afinidad que el SARS-CoV S. Además, probamos varios anticuerpos monoclonales específicos de SARS-CoV RBD publicados y descubrimos que no tienen una unión apreciable a 2019- nCoV S, lo que sugiere que la reactividad cruzada de anticuerpos puede estar limitada entre los dos RBD. La estructura de 2019-nCoV S debería permitir el rápido desarrollo y la evaluación de MCM para abordar la actual crisis de salud pública.
El nuevo coronavirus 2019-nCoV ha surgido recientemente como un patógeno humano en la ciudad de Wuhan en la provincia china de Hubei, causando fiebre, enfermedades respiratorias graves y neumonía, una enfermedad recientemente llamada COVID-19. Según la Organización Mundial de la Salud (OMS) el 16 de febrero de 2020, hubo más de 51,000 casos confirmados en todo el mundo, lo que provocó al menos 1,600 muertes. El patógeno emergente se caracterizó rápidamente como un miembro novedoso del género betacoronavirus, estrechamente relacionado con varios coronavirus de murciélago, así como con el coronavirus del síndrome respiratorio agudo severo (SARS-CoV). En comparación con el SARS-CoV, 2019-nCoV parece transmitirse más fácilmente de persona a persona, extendiéndose a múltiples continentes y dando lugar a la declaración de la OMS de Emergencia de Salud Pública de preocupación Internacional el 30 de enero de 2020.
2019-nCoV utiliza una proteína espiga (S) densamente glucosilada para ingresar a las células huésped. La proteína S es una proteína de fusión trimérica de clase I que existe en una conformación de prefusión metaestable que sufre un reordenamiento estructural dramático para fusionar la membrana viral con la membrana de la célula huésped. Este proceso se desencadena cuando la subunidad S1 se une a un receptor de la célula huésped. La unión al receptor desestabiliza el trímero de prefusión, lo que da como resultado el desprendimiento de la subunidad S1 y la transición de la subunidad S2 a una conformación estable posterior a la fusión. Para activar un receptor de la célula huésped, el dominio de unión al receptor (RBD) de S1 experimenta movimientos conformacionales tipo bisagra que ocultan o exponen transitoriamente los determinantes de la unión al receptor. Estos dos estados se conocen como conformación "abajo" y conformación "arriba", donde abajo corresponde al estado inaccesible al receptor y arriba corresponde al estado accesible al receptor, que se considera menos estable. Debido a la función indispensable de la proteína S, representa un objetivo para la neutralización mediada por anticuerpos, y la caracterización de la estructura S de prefusión proporcionaría información a nivel atómico para guiar el diseño y desarrollo de la vacuna.
Vista lateral del análisis de variabilidad 3D de CryoSPARC. Trímero 2019-nCoV S visto desde un lado, a lo largo de la membrana viral.
Vista superior del análisis de variabilidad 3D CryoSPARC. Trímero 2019-nCoV S visto desde la parte superior, hacia la membrana viral.
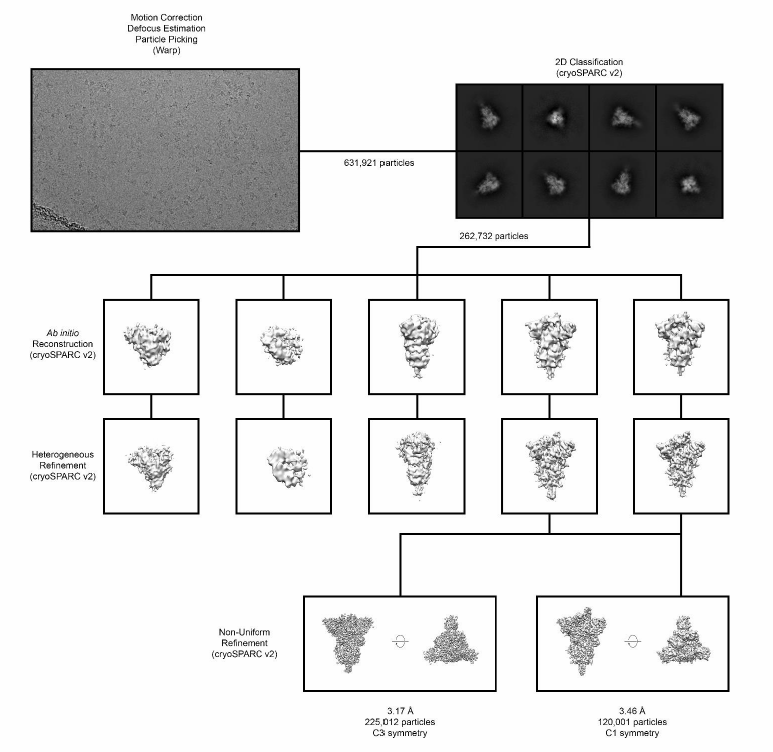
Proceso seguido hasta determinar la estructura.
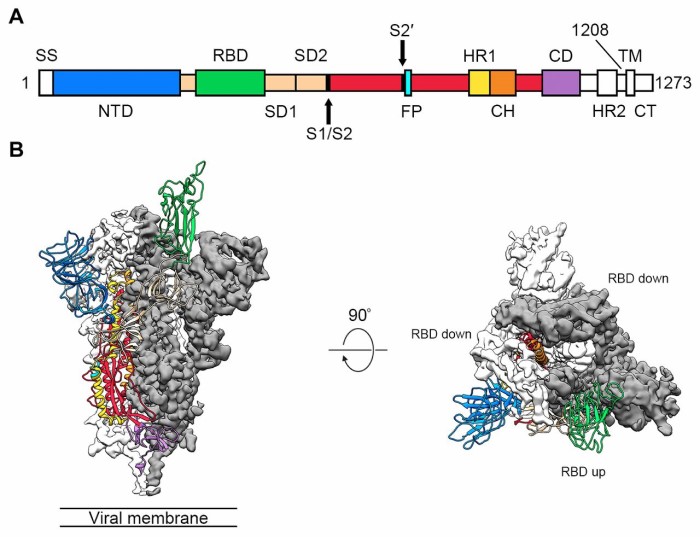
Estructura de 2019-nCoV S en la conformación de prefusión. ( A ) Esquema de la estructura primaria 2019-nCoV S, coloreada por dominio. Los dominios que se excluyeron del constructo de expresión de ectodominio (un ectodominio es el dominio de una proteína de la membrana que se extiende en el espacio extracelular (el espacio fuera de una célula). los ectodominios son por lo general las partes de las proteínas que inician contacto con superficies, lo que conduce a la transducción de señales. En SARS-CoV el ectodominio de la proteína de espiga (forma de aguja) es responsable de la unión y a la entrada en las células durante la infección) o que no pudieron visualizarse en el mapa final son de color blanco. SS = secuencia de señal, NTD = dominio N-terminal, RBD = dominio de unión al receptor, SD1 = subdominio 1, SD2 = subdominio 2, S1 / S2 = sitio de escisión de proteasa S1 / S2, S2 '= sitio de escisión de proteasa S2', FP = péptido de fusión, HR1 = repetición heptada 1, CH = hélice central, CD = dominio conector, HR2 = repetición heptada 2, TM = dominio transmembrana, CT = cola citoplasmática. Las flechas denotan sitios de escisión de proteasa. ( B ) Vistas lateral y superior de la estructura de prefusión de la proteína S 2019-nCoV con un solo RBD en la conformación ascendente. Los dos protómeros RBD-down se muestran como densidad cryo-EM en blanco o gris y el protómero RBD-up se muestra en cintas, coloreadas de acuerdo con el esquema en (A).
Ver archivo PDB en el simulador 3D.
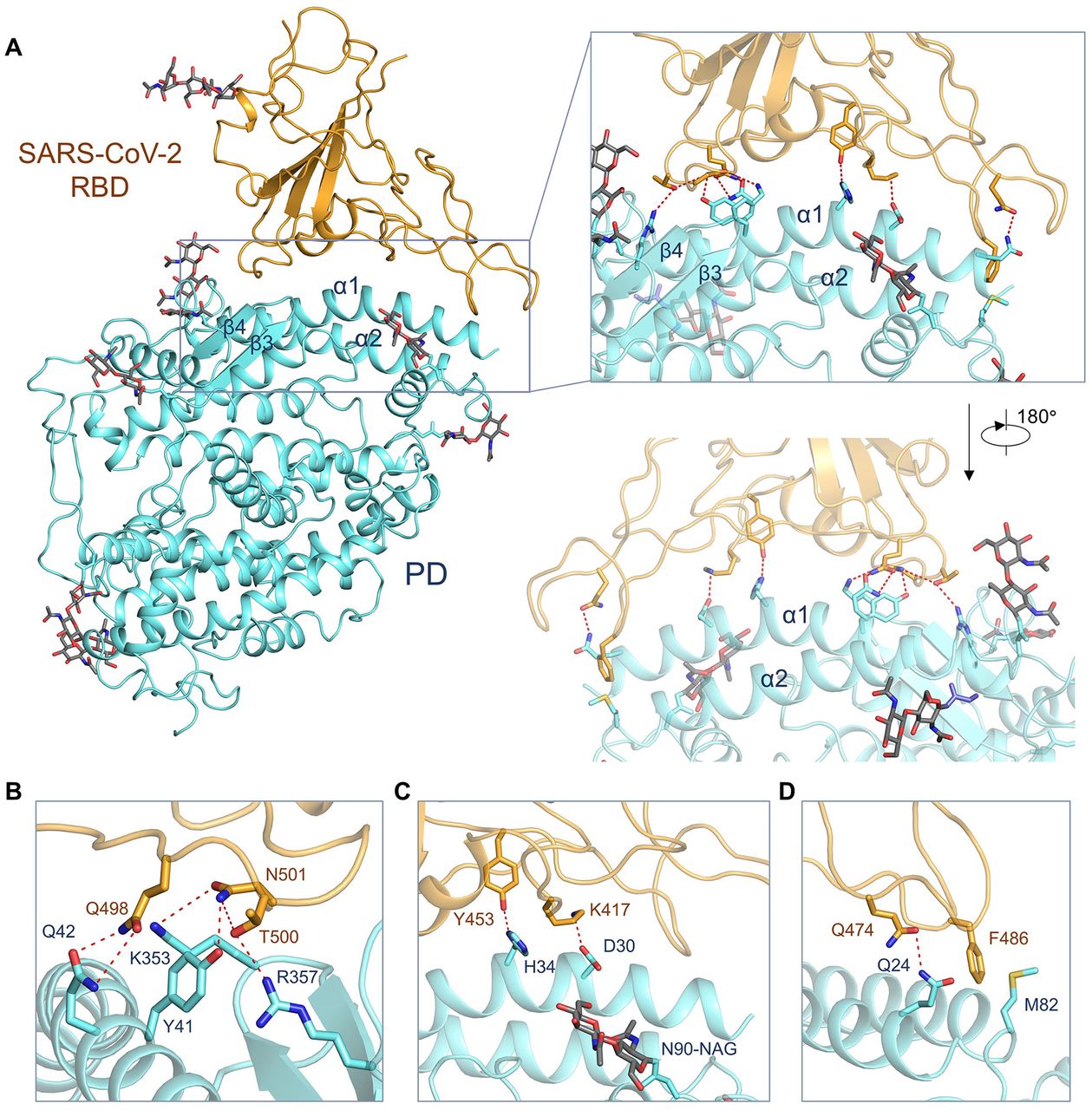
De acuerdo con la publicación de la revista Science, del 4 de marzo de 2020, un equipo de investigación determinó la estructura del complejo RBD-ACE2-B0AT1 constituído de tres partes: RBD, que corresponde a la proteína tipo espiga del virus, la enzima convertidora de angiotensina 2 -ACE2- que es el receptor celular del coronavirus del SARS (SARS-CoV) y el nuevo coronavirus (SARS-CoV-2) que está causando la grave epidemia COVID-19 y B0AT1 que es un transportador de aminoácidos.
Este descubrimiento permitirá entender una gran cantidad de aspectos bioquímicos de gran relevancia biomédica, por lo que se ha dado un gran paso en la comprensión de las bases moleculares de la infección por el coronavirus de Wuhan (COVID-19). El complejo ACE2-B0AT1 se ensambla como un dímero de heterodímeros, con el dominio tipo Collectrin (CLD) de la homo-dimerización mediadora de ACE2. El RBD es reconocido por el dominio de peptidasa extracelular (PD) de ACE2 principalmente a través de residuos polares.
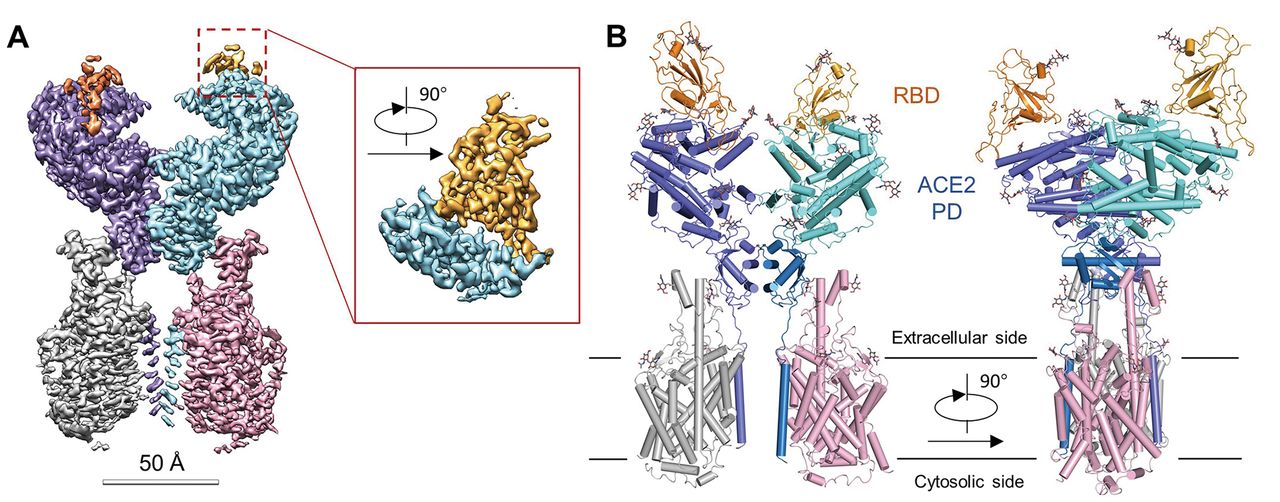
Estructura general del complejo RBD-ACE2-B0AT1. (A) Mapa de criomicroscopía electrónica del complejo RBD-ACE2-B0AT1. Izquierda: reconstrucción general del complejo ternario a 2,9 Å. Recuadro: mapa refinado enfocado de RBD. (B) Estructura general del complejo RBD-ACE2-B0AT1. El complejo está coloreado por subunidades, con el dominio de proteasa (PD) y el dominio similar a Collectrin (CLD) de color cian y azul en uno de los protómeros ACE2, respectivamente. Los restos de glicosilación se muestran como barras (crédito: Yan, R et al.).
Ver archivo PDB del complejo RBD-ACE2-B0AT1 en el simulador 3D. RBD en azul, ACE2 en rojo y B0AT1 en verde.
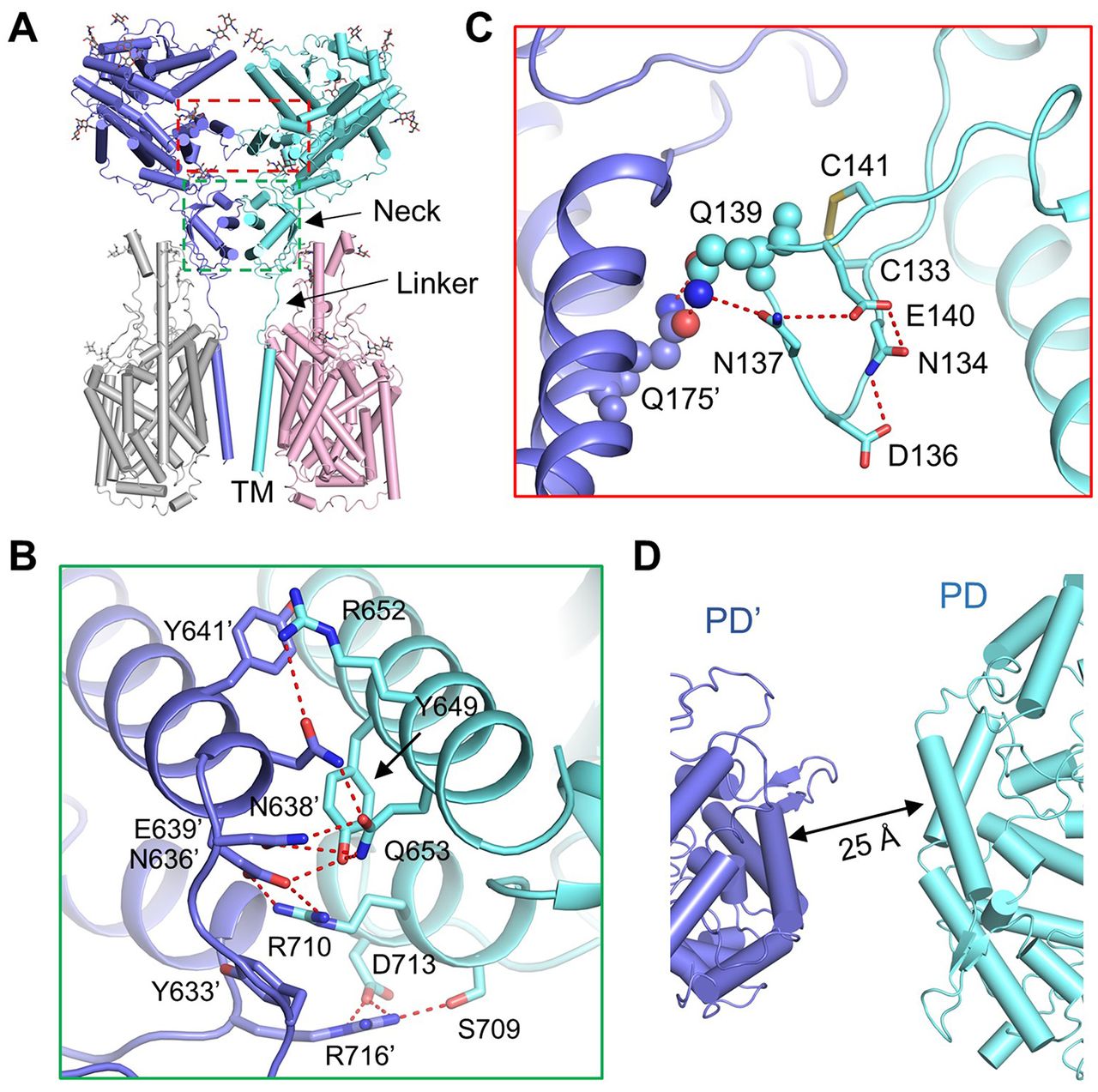
Interfaz de dimerización de ACE2. (A) ACE2 se dimeriza a través de dos interfaces, el dominio PD y el dominio Neck. (B) La interfaz dimérica primaria se encuentra entre el dominio Neck en ACE2. Las interacciones polares están representadas por líneas rojas discontinuas. (C) Una interfaz más débil entre los PD de ACE2. La única interacción es entre Gln139 y Gln175 ', que se destacan como esferas. Los residuos polares que pueden contribuir a la estabilización de Gln139 se muestran como barras. (D) Los PD ya no se contactan entre sí en estado abierto.
Ver archivo PDB del complejo ACE2-B0AT1.
Ver archivo PDB del complejo ACE2-B0AT1 en conformación abierta.
La transformación estructural entre la conformación cerrada y abierta del complejo ACE2-B0AT1.